News
GROMACS 2020 Highlights
March 2, 2020
7 min read


GROMACS 2020 was released on January 1, 2020. Patch releases may have been made since then, please use the updated versions! Here are some highlights, along with more detail in the links below!
There are several useful performance improvements, with or without GPUs, all enabled and automated by default. In addition, several new features are available for running simulations. The new features include:
Click here to view the full Release notes.
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
It is primarily designed for biochemical molecules like proteins, lipids, and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the non-bonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.

The long-term collaboration between NVIDIA and the core GROMACS developers has delivered a simulation package for biomolecular systems that performs incredibly fast. Previous GROMACS packages saw a spike in performance using GPU acceleration, but at a computational expense, especially when using multiple GPUs to run a single simulation. The new performance features available in GROMACS 2020 address these issues, and now for many typical simulations, the entire timestep can run on the GPU, avoiding CPU and PCIe bottlenecks. Inter-GPU communication operations can now operate directly between GPU memory spaces, resulting in large performance improvements.
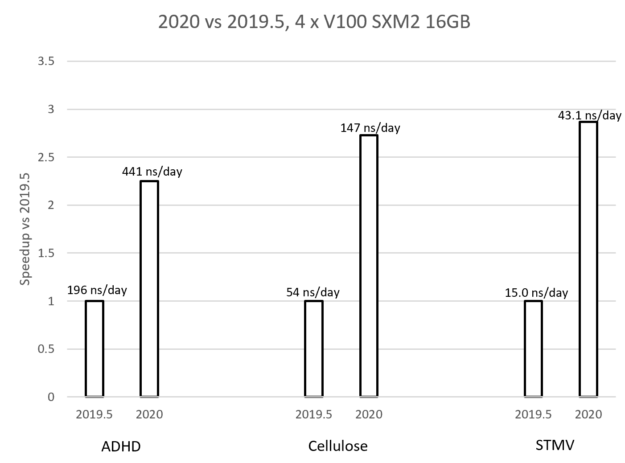
Figure 7: Comparison of GROMACS 2019 to GROMACS 2020 using three multi-GPU simulation examples.
You can read more details on the NVIDIA blog.
Download the complete full documentation to learn more: http://manual.gromacs.org/2020/manual-2020.pdf
Let us know if you have any questions about getting a system optimized for GROMACS, or upgrading your current one.

GROMACS 2020 was released on January 1, 2020. Patch releases may have been made since then, please use the updated versions! Here are some highlights, along with more detail in the links below!
There are several useful performance improvements, with or without GPUs, all enabled and automated by default. In addition, several new features are available for running simulations. The new features include:
Click here to view the full Release notes.
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
It is primarily designed for biochemical molecules like proteins, lipids, and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the non-bonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.
The long-term collaboration between NVIDIA and the core GROMACS developers has delivered a simulation package for biomolecular systems that performs incredibly fast. Previous GROMACS packages saw a spike in performance using GPU acceleration, but at a computational expense, especially when using multiple GPUs to run a single simulation. The new performance features available in GROMACS 2020 address these issues, and now for many typical simulations, the entire timestep can run on the GPU, avoiding CPU and PCIe bottlenecks. Inter-GPU communication operations can now operate directly between GPU memory spaces, resulting in large performance improvements.
Figure 7: Comparison of GROMACS 2019 to GROMACS 2020 using three multi-GPU simulation examples.
You can read more details on the NVIDIA blog.
Download the complete full documentation to learn more: http://manual.gromacs.org/2020/manual-2020.pdf
Let us know if you have any questions about getting a system optimized for GROMACS, or upgrading your current one.
