Scientific Computing
ColabDesign - Design Novel Proteins with Targeted Structure
February 9, 2024
12 min read


Advances in deep learning for protein structures have accelerated at a remarkable pace in the last few years. This has opened up a window into the myriad roles of proteins in biology and medicine and their potential to engineer-to-order capabilities displayed by proteins in nature, and more. The most obvious application for deep learning protein engineering, and likely to return the most impact in the near term, is for drug design.
We’ll demystify running protein design using Free Hallucination, Inverted Prediction, and Constrained Hallucination with RosettaFold or AlphaFold models, consolidated by Sergey Ovchinnikov and colleagues in the ColabDesign project.
AlphaFold, the AI protein folding model that burst into the life science and drug design space, is adopted in numerous labs for computer assisted drug design. Its pioneering and incomprehensible ability to accurately predict the structure of a protein when given an amino acid sequence is second to none. However, the premise of protein design is not to feed it its structural code but to obtain the code from a desired structure.
When designing a new part to be made on a 3D printer, it is unfeasible to directly type out the gcode and expect a perfect print. Instead, designing its 3D model using some CAD software of choice comes first. Then a slicer program is used to generate the gcode file. Similarly, the real promise of AlphaFold, RosettaFold, and other protein prediction models, is understanding the sequence-structure relationship for protein engineering and drug development. Design bespoke proteins with arbitrary structures and custom functionality should be the input with the amino acid sequence as the output. For that we need to not only be able to go from sequence to structure, but from structure to sequence.
There are several different approaches to generating sequences from structure, including inpainting, inverted structure prediction, and hallucination methods. Hallucination can be further subdivided into free and constrained varieties, with constrained hallucination and inverted structure prediction being closely related. We’ll use constrained hallucination in a modified version of ColabDesign to design a protein binder. But first, what are the differences and similarities between each of these techniques?
In free hallucination, the objective is to generate an accurate sequence-structure pair without regard to the functionality or ultimate structure of the protein. This means that free hallucination can give rise to protein sequences and structures that are very different from natural proteins, but they don’t fulfill a prescribed functionality.
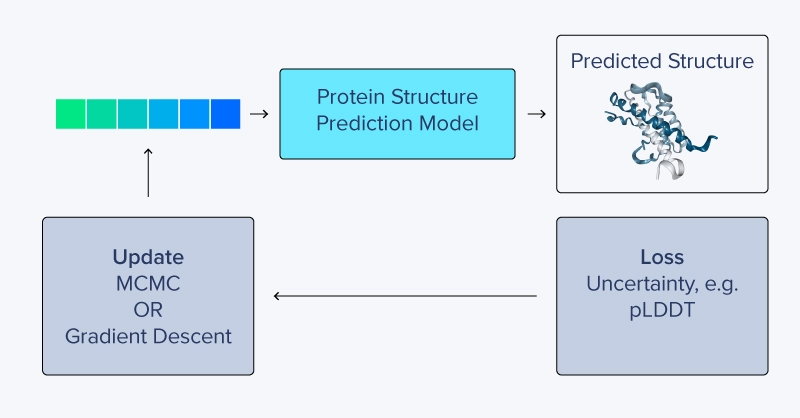
Starting from a randomly initialized sequence of amino acids, a protein structure prediction model like trRosetta, AlphaFold, or RosettaFold predicts a structure and then generates a confidence score (typically predicted Local Distance Difference Test: pLDDT). Using either gradient descent or Markov chain Monte Carlo (MCMC), the amino acid sequence can be gradually updated until a high-confidence structure is produced. Additionally, scoring terms can be included, such as Kullback-Leibler divergence from natural protein sequences as used by Anishchenko et al., to hallucinate novel and “unnatural” protein structure predictions.
The ultimate test of free hallucination is to use the resulting sequence to generate proteins in the molecular biology wet lab and solve the corresponding sequences using a technology such as cryo-EM, protein NMR, or X-Ray crystallography.
In practice, full validation in the physical lab is often not feasible for large numbers of generated protein sequences. Circular dichroism is a simpler method that can be used for crude validation of secondary structure content. However, circular dichroism only yields information about the relative secondary structure composition, not the overall structure.
Inverted structure prediction follows a similar flow to free hallucination, starting with a randomly initialized sequence of amino acids (or random logit values representing amino acids). Unlike free hallucination, inverted structure prediction aims to produce a sequence that folds to a specific structure, not just a structure prediction with high confidence. This method can be used to generate a sequence based on a target structure. Depending on the model used, the loss may be based on amino acid residue coordinates directly, or a distrogram of relative residue distances.
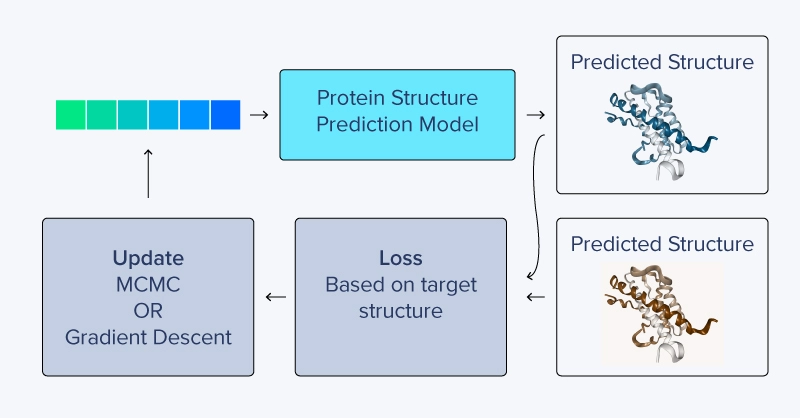
Instead of a loss function based on pLDDT, inverted structure prediction uses the difference in the predicted structure as it relates to a target structure. Updating amino acid sequences can be tricky, as the symbolic amino acid representations themselves are discontinuous variables. Sergey Ovchinnikov describes several schemes for updating the numerical representations of amino acids and found that optimizing raw logits, followed by softmax and eventually hardmax values, produced the best results. These raw values, allowed to take on very large or even negative values, are gradually replaced, residue by residue, by setting the maximum (hardmax) logit to 1.0 and others to ~0.0. This happens as the sequence is generated over multiple forward and reverse passes.
Constrained hallucination is a mix of free hallucination and inverted structure prediction. Unlike free hallucination and similar to inverted structure prediction, a target structure is defined. However, unlike inverted structure prediction, this target structure is only partially defined and accounts for only a small part of the total protein. The partial protein target may specify a functional site or structural motif of interest, or more importantly, a target binding partner.
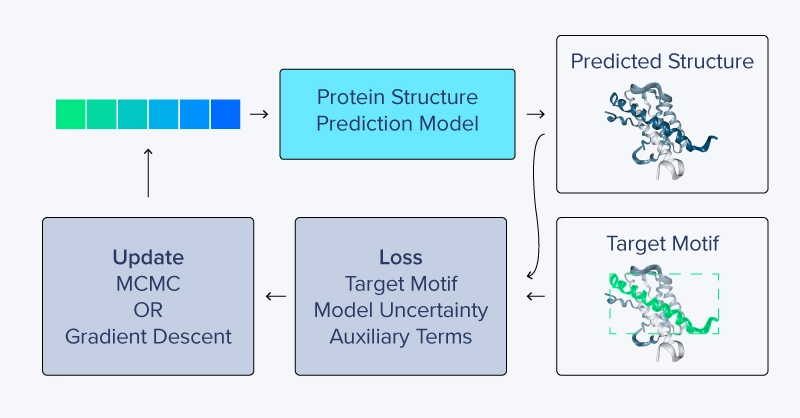
Constrained hallucination can use additional loss terms that may be problem specific. For example, in one design experiment, Wang et al. added a repulsion term to encourage the model to generate a sequence that only binds to a specific antigen-recognition area on a target antibody yet discourages non-specific and off-target interactions with the body of the antibody.
While accurate structure-sequence correspondence is an objective (and can be validated by solving the structures after expression in the lab), it is often not the objective. Constrained hallucination is well-suited for designing functional proteins, and structures with a desired functionality, such as binding to a target or enzymatic activity, can be specifically assayed in the wet lab.
Perhaps the most interesting of the design techniques discussed here is the use of inpainting. However, it may also be the most technically difficult. Hallucination schemes and inverted structure prediction can use pre-existing models trained only for structure prediction. Inpainting requires training a new model specific to the task, although it does benefit from transfer learning, starting from a model previously trained for structure prediction. Unlike hallucination methods or inverted structure prediction, inpainting requires re-training a structure prediction model with both sequence and structure inputs to fill in missing gaps.
Inpainting takes advantage of the architecture of protein structure prediction models that have both sequence and structure inputs, the latter typically used for “recycling” and iteratively improving predicted structures in multiple passes through the model. As in constrained hallucination, a portion of the structure (such as a functional site or binding partner) is specified in the model input as a starting template.
The sequence elements corresponding to the partial structure given as input to the model are filled with their corresponding amino acids, but the numerical representations of the other residue features are set to 0.0, as are the coordinates in the template structure.
For inpainting, partially specified structures and sequences serve as inputs to the model for both training and inference. As a consequence, inpainting occurs on the forward pass only, and there’s no need for comparing clever tricks for updating sequence logits via back-propagation at inference time.
ColabDesign was developed in part as a relatively easy tool for protein design using Google Colaboratory, but it is also perfectly suited for local use. To get started, you’ll first need to download the repository and install some dependencies. First, clone the modified repo (forked from the original) using git.
# clone the repository
git clone git@github.com:riveSunder/LocalColabDesign.git
cd LocalColabDesign
Using a virtual environment to manage dependencies is recommended. The preferred manager is virtualenv, but you may adjust the following commands to use your virtual environment manager of choice.
# set up a virtual environment
virtualenv ./local_design --python=python3.8
source ./local_design/bin/activate
# install jupyter (for notebooks)
pip install jupyter==1.0.0
# install ColabDesign
pip install -e .
Next, you’ll need to download AlphaFold parameters from Google.
# download and decompress af params
mkdir params
wget
https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar
tar -xf alphafold_params_2022-12-06.tar -C params
You’ll probably want to run ColabDesign with the help of your GPU, if possible. The correct version of JAX for this CUDA version (CUDA 11) can be installed with the following command:
pip install --upgrade "jax[cuda11_local]" -f \
https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
If you have CUDA 12, you can replace ‘cuda11_local’ with ‘cuda12_pip’. Check the JAX repository documentation to match your hardware to the right install options.
That’s all you should need to design your own protein binders.
You can now type ‘jupyter notebook’ at the command line, navigate to af/local_binder_design.ipynb, and select Kernel>Restart Kernel and Run All Cells.
The first cell checks for a params folder containing AlphaFold parameters and then imports dependencies. Next, you’ll find a cell for preparing to run the design process by specifying the target PDB id and chain, and the desired binder length. “1YCR” is the PDB id for Mdm2 bound to P53.
clear_mem()
af_model = mk_afdesign_model(protocol="binder")
af_model.prep_inputs(pdb_filename=get_pdb("1YCR"), chain="A", binder_len=128)
The execution cell resets the model and calls design_3stage, which takes 3 numerical arguments specifying how many steps to run in each stage.
Constrained hallucination is an iterative process that uses gradient descent, but aims to optimize the numerical representation of an amino acid sequence instead of optimizing model parameters. The 3 stages refer to 3 different ways of treating the sequence representation during optimization, which when applied sequentially, can specify the number of steps to run optimization in each stage.
af_model.restart()
af_model.design_3stage(100,100,10)
Stage 1 starts by applying gradient descent to the sequence logits directly, freely allowing these values to range from very large to even negative magnitudes. This stage gradually switches to gradient descent on softmax(logits).
Stage 2 starts with softmax(logits) and gradually switches to hardmax(logits).
Stage 3 finalizes the design on hardmax(logits). This progression from raw logits to hardmax(logits) was found by Ovchinnikov et al. to produce better results than raw logits, softmax(logits), or hardmax(logits) alone.
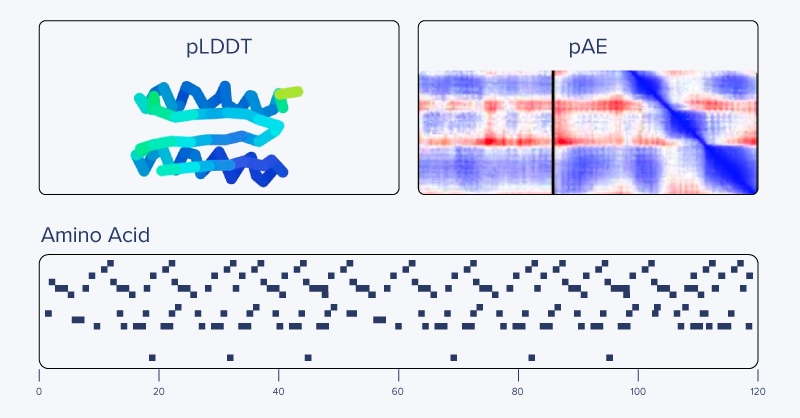
The constrained hallucination optimization process does take time, which amounts to roughly an hour or so on our machine. If all goes well, you’ll be rewarded with a new protein binder design, and you can investigate the results by visualizing the sequence optimization process and the resulting structure coded by model-predicted uncertainty in pLDDT.
Protein structure prediction was revolutionized by DeepMind’s AlphaFold project at CASP14 in 2020. Since then, several peer models have been developed, including most famously the RosettaFold project from David Baker at the University of Washington and colleagues.
Sergey Ovchinnikov (soon to be at MIT) and colleagues developed ColabDesign to facilitate protein engineering with AlphaFold/RoseTTAFold models. In this article, we discussed methods for using gradient descent on sequence logit values for protein design via free hallucination, inverted protein structure prediction, and constrained hallucination as well as the forward-pass-only method of inpainting. We also used a modified version of ColabDesign to run constrained hallucination to design a protein binder for Mdm2, which is a negative regulator of P53 and a potential target for treating cancer.
Protein therapeutics are the most obvious application, but improving health and treating disease is only the tip of the AI protein engineering iceberg. Designing custom and arbitrary proteins with capable machine learning models may ultimately have a much greater impact. In nature, proteins play all kinds of roles including chemical catalysis, signaling, sensing, building robust structures at multiple scales, and others. In future developments, protein design powered by deep learning could be used to design better materials and improved catalysts for a wide variety of biotechnological processes.
Advances in deep learning for protein structures have accelerated at a remarkable pace in the last few years. This has opened up a window into the myriad roles of proteins in biology and medicine and their potential to engineer-to-order capabilities displayed by proteins in nature, and more. The most obvious application for deep learning protein engineering, and likely to return the most impact in the near term, is for drug design.
We’ll demystify running protein design using Free Hallucination, Inverted Prediction, and Constrained Hallucination with RosettaFold or AlphaFold models, consolidated by Sergey Ovchinnikov and colleagues in the ColabDesign project.
AlphaFold, the AI protein folding model that burst into the life science and drug design space, is adopted in numerous labs for computer assisted drug design. Its pioneering and incomprehensible ability to accurately predict the structure of a protein when given an amino acid sequence is second to none. However, the premise of protein design is not to feed it its structural code but to obtain the code from a desired structure.
When designing a new part to be made on a 3D printer, it is unfeasible to directly type out the gcode and expect a perfect print. Instead, designing its 3D model using some CAD software of choice comes first. Then a slicer program is used to generate the gcode file. Similarly, the real promise of AlphaFold, RosettaFold, and other protein prediction models, is understanding the sequence-structure relationship for protein engineering and drug development. Design bespoke proteins with arbitrary structures and custom functionality should be the input with the amino acid sequence as the output. For that we need to not only be able to go from sequence to structure, but from structure to sequence.
There are several different approaches to generating sequences from structure, including inpainting, inverted structure prediction, and hallucination methods. Hallucination can be further subdivided into free and constrained varieties, with constrained hallucination and inverted structure prediction being closely related. We’ll use constrained hallucination in a modified version of ColabDesign to design a protein binder. But first, what are the differences and similarities between each of these techniques?
In free hallucination, the objective is to generate an accurate sequence-structure pair without regard to the functionality or ultimate structure of the protein. This means that free hallucination can give rise to protein sequences and structures that are very different from natural proteins, but they don’t fulfill a prescribed functionality.
Starting from a randomly initialized sequence of amino acids, a protein structure prediction model like trRosetta, AlphaFold, or RosettaFold predicts a structure and then generates a confidence score (typically predicted Local Distance Difference Test: pLDDT). Using either gradient descent or Markov chain Monte Carlo (MCMC), the amino acid sequence can be gradually updated until a high-confidence structure is produced. Additionally, scoring terms can be included, such as Kullback-Leibler divergence from natural protein sequences as used by Anishchenko et al., to hallucinate novel and “unnatural” protein structure predictions.
The ultimate test of free hallucination is to use the resulting sequence to generate proteins in the molecular biology wet lab and solve the corresponding sequences using a technology such as cryo-EM, protein NMR, or X-Ray crystallography.
In practice, full validation in the physical lab is often not feasible for large numbers of generated protein sequences. Circular dichroism is a simpler method that can be used for crude validation of secondary structure content. However, circular dichroism only yields information about the relative secondary structure composition, not the overall structure.
Inverted structure prediction follows a similar flow to free hallucination, starting with a randomly initialized sequence of amino acids (or random logit values representing amino acids). Unlike free hallucination, inverted structure prediction aims to produce a sequence that folds to a specific structure, not just a structure prediction with high confidence. This method can be used to generate a sequence based on a target structure. Depending on the model used, the loss may be based on amino acid residue coordinates directly, or a distrogram of relative residue distances.
Instead of a loss function based on pLDDT, inverted structure prediction uses the difference in the predicted structure as it relates to a target structure. Updating amino acid sequences can be tricky, as the symbolic amino acid representations themselves are discontinuous variables. Sergey Ovchinnikov describes several schemes for updating the numerical representations of amino acids and found that optimizing raw logits, followed by softmax and eventually hardmax values, produced the best results. These raw values, allowed to take on very large or even negative values, are gradually replaced, residue by residue, by setting the maximum (hardmax) logit to 1.0 and others to ~0.0. This happens as the sequence is generated over multiple forward and reverse passes.
Constrained hallucination is a mix of free hallucination and inverted structure prediction. Unlike free hallucination and similar to inverted structure prediction, a target structure is defined. However, unlike inverted structure prediction, this target structure is only partially defined and accounts for only a small part of the total protein. The partial protein target may specify a functional site or structural motif of interest, or more importantly, a target binding partner.
Constrained hallucination can use additional loss terms that may be problem specific. For example, in one design experiment, Wang et al. added a repulsion term to encourage the model to generate a sequence that only binds to a specific antigen-recognition area on a target antibody yet discourages non-specific and off-target interactions with the body of the antibody.
While accurate structure-sequence correspondence is an objective (and can be validated by solving the structures after expression in the lab), it is often not the objective. Constrained hallucination is well-suited for designing functional proteins, and structures with a desired functionality, such as binding to a target or enzymatic activity, can be specifically assayed in the wet lab.
Perhaps the most interesting of the design techniques discussed here is the use of inpainting. However, it may also be the most technically difficult. Hallucination schemes and inverted structure prediction can use pre-existing models trained only for structure prediction. Inpainting requires training a new model specific to the task, although it does benefit from transfer learning, starting from a model previously trained for structure prediction. Unlike hallucination methods or inverted structure prediction, inpainting requires re-training a structure prediction model with both sequence and structure inputs to fill in missing gaps.
Inpainting takes advantage of the architecture of protein structure prediction models that have both sequence and structure inputs, the latter typically used for “recycling” and iteratively improving predicted structures in multiple passes through the model. As in constrained hallucination, a portion of the structure (such as a functional site or binding partner) is specified in the model input as a starting template.
The sequence elements corresponding to the partial structure given as input to the model are filled with their corresponding amino acids, but the numerical representations of the other residue features are set to 0.0, as are the coordinates in the template structure.
For inpainting, partially specified structures and sequences serve as inputs to the model for both training and inference. As a consequence, inpainting occurs on the forward pass only, and there’s no need for comparing clever tricks for updating sequence logits via back-propagation at inference time.
ColabDesign was developed in part as a relatively easy tool for protein design using Google Colaboratory, but it is also perfectly suited for local use. To get started, you’ll first need to download the repository and install some dependencies. First, clone the modified repo (forked from the original) using git.
# clone the repository
git clone git@github.com:riveSunder/LocalColabDesign.git
cd LocalColabDesign
Using a virtual environment to manage dependencies is recommended. The preferred manager is virtualenv, but you may adjust the following commands to use your virtual environment manager of choice.
# set up a virtual environment
virtualenv ./local_design --python=python3.8
source ./local_design/bin/activate
# install jupyter (for notebooks)
pip install jupyter==1.0.0
# install ColabDesign
pip install -e .
Next, you’ll need to download AlphaFold parameters from Google.
# download and decompress af params
mkdir params
wget
https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar
tar -xf alphafold_params_2022-12-06.tar -C params
You’ll probably want to run ColabDesign with the help of your GPU, if possible. The correct version of JAX for this CUDA version (CUDA 11) can be installed with the following command:
pip install --upgrade "jax[cuda11_local]" -f \
https://storage.googleapis.com/jax-releases/jax_cuda_releases.html
If you have CUDA 12, you can replace ‘cuda11_local’ with ‘cuda12_pip’. Check the JAX repository documentation to match your hardware to the right install options.
That’s all you should need to design your own protein binders.
You can now type ‘jupyter notebook’ at the command line, navigate to af/local_binder_design.ipynb, and select Kernel>Restart Kernel and Run All Cells.
The first cell checks for a params folder containing AlphaFold parameters and then imports dependencies. Next, you’ll find a cell for preparing to run the design process by specifying the target PDB id and chain, and the desired binder length. “1YCR” is the PDB id for Mdm2 bound to P53.
clear_mem()
af_model = mk_afdesign_model(protocol="binder")
af_model.prep_inputs(pdb_filename=get_pdb("1YCR"), chain="A", binder_len=128)
The execution cell resets the model and calls design_3stage, which takes 3 numerical arguments specifying how many steps to run in each stage.
Constrained hallucination is an iterative process that uses gradient descent, but aims to optimize the numerical representation of an amino acid sequence instead of optimizing model parameters. The 3 stages refer to 3 different ways of treating the sequence representation during optimization, which when applied sequentially, can specify the number of steps to run optimization in each stage.
af_model.restart()
af_model.design_3stage(100,100,10)
Stage 1 starts by applying gradient descent to the sequence logits directly, freely allowing these values to range from very large to even negative magnitudes. This stage gradually switches to gradient descent on softmax(logits).
Stage 2 starts with softmax(logits) and gradually switches to hardmax(logits).
Stage 3 finalizes the design on hardmax(logits). This progression from raw logits to hardmax(logits) was found by Ovchinnikov et al. to produce better results than raw logits, softmax(logits), or hardmax(logits) alone.
The constrained hallucination optimization process does take time, which amounts to roughly an hour or so on our machine. If all goes well, you’ll be rewarded with a new protein binder design, and you can investigate the results by visualizing the sequence optimization process and the resulting structure coded by model-predicted uncertainty in pLDDT.
Protein structure prediction was revolutionized by DeepMind’s AlphaFold project at CASP14 in 2020. Since then, several peer models have been developed, including most famously the RosettaFold project from David Baker at the University of Washington and colleagues.
Sergey Ovchinnikov (soon to be at MIT) and colleagues developed ColabDesign to facilitate protein engineering with AlphaFold/RoseTTAFold models. In this article, we discussed methods for using gradient descent on sequence logit values for protein design via free hallucination, inverted protein structure prediction, and constrained hallucination as well as the forward-pass-only method of inpainting. We also used a modified version of ColabDesign to run constrained hallucination to design a protein binder for Mdm2, which is a negative regulator of P53 and a potential target for treating cancer.
Protein therapeutics are the most obvious application, but improving health and treating disease is only the tip of the AI protein engineering iceberg. Designing custom and arbitrary proteins with capable machine learning models may ultimately have a much greater impact. In nature, proteins play all kinds of roles including chemical catalysis, signaling, sensing, building robust structures at multiple scales, and others. In future developments, protein design powered by deep learning could be used to design better materials and improved catalysts for a wide variety of biotechnological processes.
