Artificial Intelligence
Deep Learning Meets Molecular Dynamics: "Predicting Correctness of Protein Complex Binding Operations"- An Interview With Stanford Students
July 19, 2019
20 min read


For this blog post, James Montantes from Exxact sat down with some Stanford CS students to discuss their project "Predicting Correctness of Protein Complex Binding Operations" that was presented at the CS229 poster session on December 2018. This particular poster session was of particular interest in that it shows an interesting application of deep learning for bio-chemistry, while comparing more traditional machine learning methods such as SVM to attack this particular problem. Students; Sarah Gurev, Nidhi Manoj, and Kaylie Zhu sat down with James for a quick 30-minute discussion at the bustling Coupa Cafe on the Stanford campus to discuss some of the technical challenges of the project.
James Montantes: Thanks for giving me the opportunity to talk about your project, it was very interesting. Typically, I go to these poster sessions whenever I get the chance, I think it's a great opportunity to see a lot of interesting applications for machine learning. Given its recent popularity in the past several years, there's some real industry use cases applying these algorithms to the GPU, solving real problems. At the university level, I find these poster sessions offer a lot of interesting use cases. Which brings me to your poster, I thought it was interesting because you're going after a unique application, which would be molecular dynamics. Given the field I’m working in, I felt it would be a great fit... Having said all that, tell me a little bit about yourselves and what you're working on, your background and interests etc.
Sarah: I'm a junior majoring in computer science, in the Biocomputation track. I've been interested in both biology and computer science for a while. I started off working in a pure bio lab doing cancer biology research, and then moved to a structural biology computational lab, the Levitt Lab, working on molecular dynamics which is how I was interested in our project. For lab, I'm modeling chemokine-chemokine receptor interaction using molecular dynamics and analyzing those to understand the basis of affinity of the interaction.
JM: Thank you. Kaylie?
Kaylie: Yeah, I’m a junior in the Symbolic systems major (AI concentration) and also co-term in CS with AI depth. I've been working previously with the Stanford AI group with deep learning applications regarding health care. Since then I've just been interested in deep learning and different kinds of applications in computer vision, for example, food recognition, satellite imagery, map technologies, et cetera. I really enjoy discovering new uses of deep learning and in this case, we've applied it to bio-molecular dynamics which is a new field for me and I thought that was very exciting.
JM: Ok, thank you, Nidhi?
Nidhi: I'm a senior studying computer science with a focus in AI. And, co-term student so it's the master's program here that’s also in CS with a focus in artificial intelligence, again. In terms of research interests, Kaylie and I were part of the same lab. That's how we actually met. It's the AI plus health care lab in the Stanford Machine Learning Group under Andrew Ng. And that is, as Kaylie articulated, deep learning, focused on health care problems, usually in radiology or pathology. Now I'm working with Fei-Fei Li in the AI plus health care sector of her lab that's also in the Stanford Machine Learning Group. During the summers I enjoy interning just to try to get a sense of both research and industry from both sides of it, to see the application side as well. And I guess, we were all kind of interested in the intersection of artificial intelligence and biology when we were taking a class together. All our backgrounds coming together was interesting to work on the bio-computation project.
JM: Great. Thank you, now this seems like a very specific problem, specific to finding protein docking orientations and what not… How did you come across this? How did you feel this problem could be solved with machine learning? And what was the inception of this idea?
Sarah: I talked to a postdoc in my lab, João Rodrigues, who’d made the HADDOCK models from Docking Benchmark data. So when I came to him looking for a dataset for CS229, he suggested this. It’s a good application for Machine Learning because there's so many protein complexes that we'll just never have their structures experimentally determined, but scientists would like to be able to use those protein complex structures to advance their understanding. So, it's useful to be able to use some techniques to determine what simulated models are the proper models.
JM: Excellent… So now going back to machine learning, what kind of machine learning, or deep learning tools or models did you use to solve your problems? How did that come about? I saw in your paper you used scikit-learn to use some of the regression testing for the SVM (Support Vector Machine) etc.
Nidhi: For the SVM implementation, which is our baseline for both regression and classification, we were using scikit-learn, which just really simplifies the implementation of the model. Then we transitioned into a deep learning, 3D CNN (Convolutional Neural Network) resident model that Kaylie can talk a little bit more about.
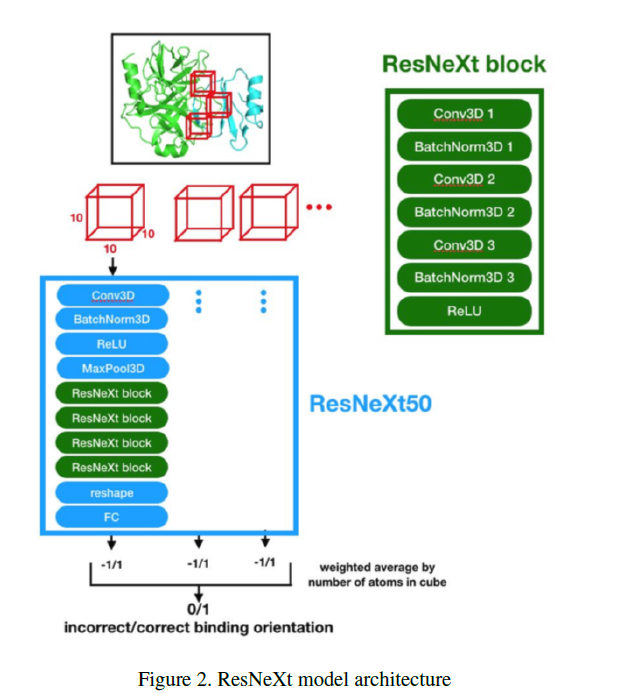
Kaylie: For the 3D CNN we used PyTorch, which is a super useful library for deep learning. All three of us prefer it over TensorFlow, so that's what we chose. Also, we built on top of a model that was built by other people for other uses called ResNeXt. It's a 3D convolutional neural network that's used for 3D data and commonly used for video recognition. In our case, it made more sense, because our cubes were three-dimensional and we needed to capture spatial information. It just works extremely well in capturing the spatial context of molecules and the atoms architecture. It was super convenient for us to build because this library has built-in ways to tune learning rates and batch sizes and we could experiment with different model depths; for example, we tried ResNet 50 & 101, and we found some interesting results.
JM: Ok, I know the ResNet is a very popular model, and it's widely used. So the ResNeXt is kind of a newer architecture?
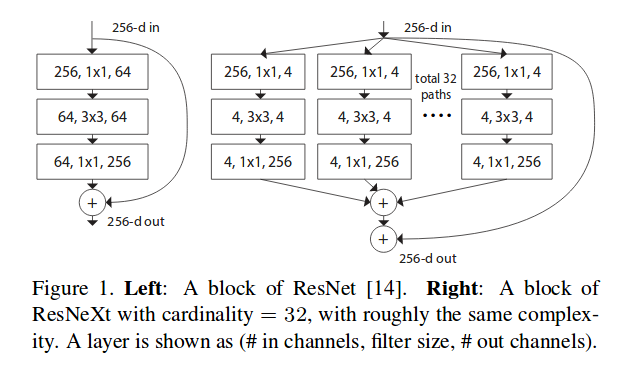
Image source: Aggregated Residual Transformations for Deep Neural Networks, Saining Xie, et al. 2017
Nidhi: It's better for 3D data
JM: I see, it lends itself to 3D spatial data.
Nidhi: Yes, and just for context, basically what we're trying to say is the protein complex equals a 3D representation, that's why a 2D model probably wouldn't work as well. The main reason you use ResNet or ResNeXt is residual connections. It's just easier for training.
Kaylie: ResNeXt, it has the advantages carried over from ResNet of being highly modularized and skipping the layers in order to better perpetrate the network flow of information of the gradients. But it also has a new dimension called cardinality, basically it introduces group convolutions and controls the size of the set of more complex transformations, an essential factor in addition to (and in fact more effective than) the dimensions of depth and width (when it comes to gaining accuracy, as shown by our experiments).
JM: Do you know if there's a limit of how many dimensions it can spread out?
Kaylie: So ResNeXt is technically for 3D data, but people can enhance it in order to adapt for more numbers of dimensions as well. But for our case, it made sense to apply for 3D.
JM: When when you implement a large ResNet model or you're training on a huge data set, it can take a long time. How about your model and dataset using ResNeXt, does it take a long time to train? Was it very data intensive?

Sarah: The data input is relatively small compared to large images with many pixels. Our data was only 10X10X10. Each voxel of that is a 1X1X1 cubic angstrom box, about the size to indicate whether one atom was present. We split our protein into these 10X10X10 cubes along the interface between the two proteins and each voxel has a zero there no atom was present or else a specific number depending on the atom. This allowed us to have a 3D representation of the complex, but where inputs were all the same size.
Nidhi: Also, for context, yes, it does take a long time to train. We were using a GPU from the Stanford Levitt Lab, so that made it realistic to train under our time constraints.
Kaylie: And ResNeXt 101 was definitely slower to train than (ResNeXt) 50.
Sarah: We got our results within a day, so it wasn't too long, and was within the scope of our project.
JM: So you use this convolutional neural network, this interesting ResNeXt model, but you also tested it against the SVM regression and classification. Is that sort of a benchmark in that you wanted to see how well it performed against traditional machine learning algorithm? Was that idea behind it or you wanted to see "do we really need deep learning," or…?
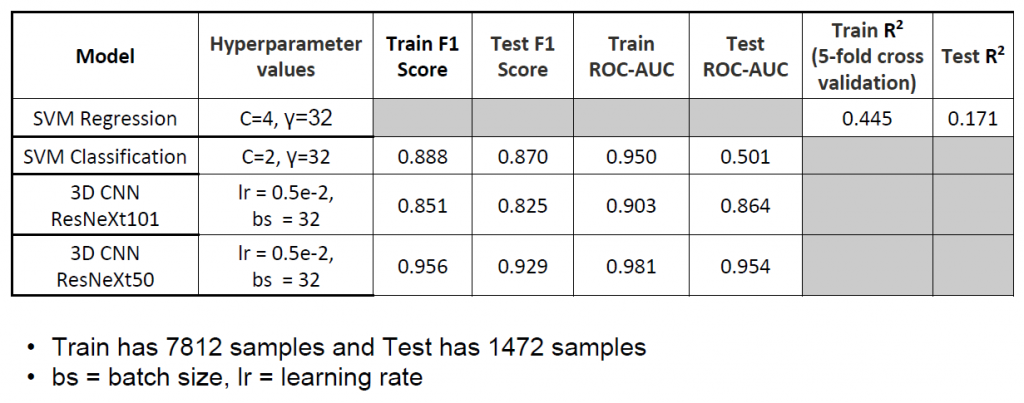
Nidhi: So, usually, these projects, we want to start off with a baseline to build off of and build more complex models. For our baseline we decided to do SVM regression and classification as articulated in our paper. We chose those because we read in previous work that SVMs tend to have good results in this. And the features we used, also mentioned in our paper, were solvation energy, van der Waals, buried surface area, and electrostatics, that has been shown to have good results in the past. We started off with that, and then we made use of the computational power of deep learning networks and that's how we started working on 3D CNNs.
Kaylie: I think if you look at our paper, the accuracy of the training results are pretty high, but the tests were a lot lower. So that's basically saying that there is some significant amount of overfitting done by the SVMs. This was tackled a lot better in our 3D CNN model, which was able to learn the more complicated relationships within the dataset rather than just overfitting to the training data. We saw that after significant tuning, it was actually able to learn and generalize to the test sets. That was one of the significant improvements in using deep learning versus the traditional machine learning model. But sometimes, in other cases, traditional machine learning is enough to produce sufficient results. Just in this case, it wasn’t sufficient to capture all the information and data complexities as well.

Nidhi: Just for some context, usually in a machine learning project, you want to split up your data into training and test. So, you train essentially on the training set, and what Kaylie was saying is when you overfit, it's kind of understanding those examples ‘too well’, therefore when you give it new data in the test set, it's not able to generalize. Basically, the SVM model was a little too simple. It was just overfitting on the training set, so that's why we resorted to CNN, which had better results in the test set, which is what you want.

Sarah: It was important that we split by complexes when we were dividing our data into training, validation, and test sets. There's 10000 HADDOCK dockings for each of the 135 complexes. We split by complex, so that when we are testing our model, it hasn’t already been trained on simulations from the same complex, preventing bias.
Nidhi: You can think of it as our problem is essentially: if given a new protein complex, how would our model be able to perform on that? So that's why your performance in the test set, and making sure you have complexes that your attributes, after you've trained this, after you've built a great model, it can perform well.
JM: Okay. That's great. That's good information. You mentioned that you gave with your training data sets, kind of a more positive results? What was that?
Sarah: There are significantly more bad models than good models, so we needed to reduce the number of negative models. Because we do 10000 dockings for each complex and the orientation is random, only a few of them are going to be proper, and by proper, I mean the RMSD, the root-mean-square deviation from the true protein complex, is small. So, only a few of these random orientations will be close to true. We found it necessary to significantly undersample the bad examples so that we had a comparable number of good examples and bad examples to train with.
Nidhi: Think about how proteins could combine, there's like a million ways they could combine. The reason you want an equal number of positive and negative is just that will help a lot with training. When you have class imbalance between positive and negative samples, it's just a lot harder for the model to learn. Yeah.
JM: I see. Fair enough. I also noticed in your paper, you mentioned some existing tools that maybe don't specifically go after the same problem, like AtomNet, that's commercially available, is it not? I was just curious had you had any experience with any of these: There's ENZYNET, which is a two-layer 3D convolutional neural network for enzyme.
Sarah: We've never used any of these models, but we're encouraged to find background research so we can understand what makes our project different from what's been done in the past. We find the pros and cons of the methods that have been used before.
Nidhi: You look to see what's happened before and want to learn from that, and guide your future research, like what kind of models are you can use.

JM: I see some of the ones that you're using like SVM models, like ProQdoc, ZRANK.
Nidhi: Yeah.
Sarah: The past models helped us also choose our parameter values for the SVM, because we do a grid search over a specific range. So we also used that same range as the past models because they are likely to be appropriate.
JM: That makes sense. This ties into a question that maybe you could help with...let's say you’ve worked in Life Sciences for years and years and done a lot of molecular dynamics work, but maybe just hearing about some of this ‘deep learning stuff’, or don’t really know how to really approach or implement some of these experiments. What kind of advice would you give them? How would they basically want to get started in it? Please explain in your own words.
Sarah: If they have a specific project in mind it's definitely a good idea to see what's already been done. They can also repeat what's already been done just for themselves, so they can learn the steps of what to take, and use tools like scikit-learn to do a basic experiment and understand the process.
Kaylie: You can also look at what kind of evaluation metrics most people, or other published work in the field have been evaluating models on because that's important. What aspects of the model performance do people actually care about, or what does the industry actually care about. Also, I think, if they have less background, they could look into transfer learning, which is basically transferring learned knowledge from a network of a different but similar problem to a new problem, and that requires a lot less technical depth, but it sometimes actually does amazingly well with some type of parameter tuning. So, if they are interested in image recognition or image captioning. There's a lot of networks that have been built by other people and if they’re are similar enough and with pre-trained weights, then they could load pre-trained weights, then either modify the last few layers or just freeze or retrain layers in order to adapt it to their problem. At times, it works exceedingly well.
Nidhi: I would say, I think if you want to get more...If you're interested in ML and want to learn a little bit more, I would start off by taking Coursera classes or reading up on MOOCs
JM: (Somewhat jokingly): How about CS 229 at Stanford?
Nidhi: Exactly. But maybe something a little easier than 229.
Kaylie: (CS) 230
Nidhi: Yeah, 230, 229, they're all great. I think that provides some necessary foundation of underlying concepts that's necessary. Otherwise, I think, I guess jumping into research papers can sometimes be a little daunting and just a lot of technical terms and concepts that are a little hard to grasp without any previous understanding. Then I think once you read up on papers, just try working on some projects. Make use of libraries, like Sarah said. Machine learning is a community, so you want to build off of the amazing work that's been done before in the field for whatever you work on. Also, all the results you have, contribute it back so that other people can build off it and keep pushing the field forward.
JM: Yeah, I've noticed that myself with the machine learning community. A lot of the software, and a lot of the papers are all open for anyone to read and download and try the code.
Nidhi: Exactly. Open source code.
JM: It's not always like that with some other areas. I've seen that trend as well, it's really welcoming...Going back to the experiments, what was the most time-consuming process or what was the thing that really frustrated you the most in this?
Kaylie: Data Prep!
Sarah: There's a lot of data processing, to convert our starting data into a usable form that can go into a deep learning model. We started with a PDB file, which is basically just a list of positions for every atom.
Nidhi: Protein data bank.
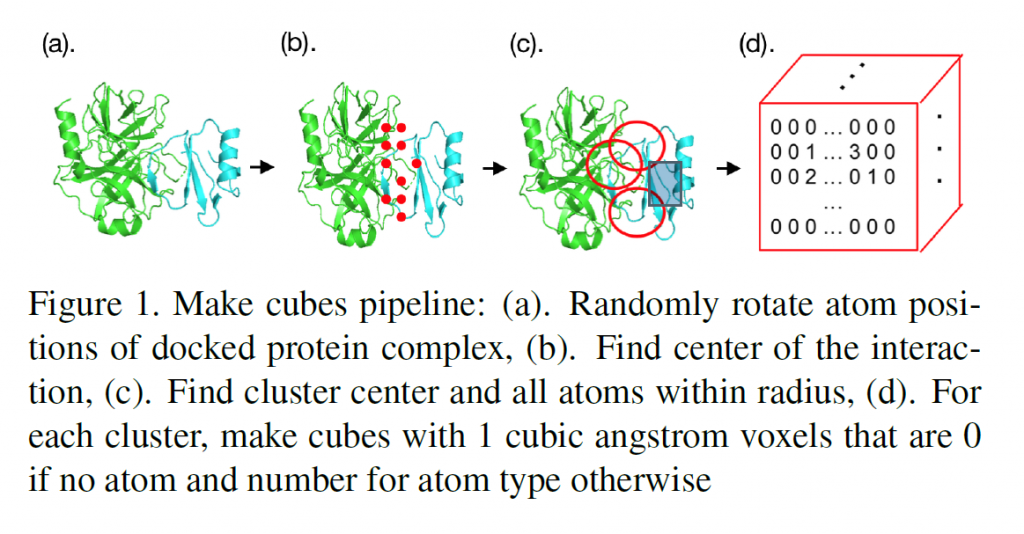
Sarah: You have all the atoms and you have the position in 3D space of those atoms, and you want to somehow turn that giant list into a 3D representation of a small portion of that complex that represents the interface. So, the way I ended up doing this was looking for pairs of amino acids, where one was on one protein and one was on the other. I’d then find the center of mass of each of those pairs and clustering those center of masses. I’d take the center of each cluster to be the center of the cube entering our model and then building the cubes around that. It was just a lot of work that went into turning this list of all these atoms positions into a usable form.
JM: And then that would be interpreted by the neural network?
Sarah: Some tools that we used were PyMol for visualization and MDTraj. It was important that we randomly rotate each complex because we didn’t want a specific orientation to influence the model. Since we have multiple orientations that are put in the model for each complex, we don’t want all those orientations to be the same.
JM: When you're setting the parameters for the network, how did you go about that? Was it established on things that have been done before that you thought would work well? Was it a lot of trial and error? How did selecting those come about?
Kaylie: So, there's two parts to it. From related work you can kind of ball park where the hyper parameters would be and then narrow down by experience and empirically. For example, ADAM optimizer tends to do well with its default parameters, you don't usually need to tune it as it doesn't produce as much effect. I think the first thing to tune would be learning rate, we usually default with 1e−3. But for us, we were like, "Oh this is actually too big," it was very clear once you look at how the training error has been decreasing over the epochs, you're clearly seeing that it's actually missing the optimum point and it was jumping around a lot so we decided to decrease the initial learning rate.
JM: Ok, so don’t mess with ADAM optimizer, and tune the learning rate starting at 1e−3.
Kaylie: We also reduced learning rate on plateau, which is a very handy trick that we found to be useful in other projects as well. We just decided to use it basically once the validation loss plateaus, we reduce the learning rate in order to find more fine-grained way of finding the optimum point. And rather than risk either missing it or not reaching there in time.
Kaylie: Also, things that we try out are batch sizes, which is significant. There's a lot of empirical testing to it, for example, you usually try out 64, 32, 16, et cetera, depending on your memory constraints and what works best for the problem. I guess there's a lot of ‘rule of thumb’ you can follow and things you can learn from experience and also from other people’s work. But there's a lot of empirical testing, which I think is actually really inevitable in work like this.
JM: Excellent. Okay, so I think that pretty much goes over most of the technical questions. Was there anything else you want to add or points of interest? I know you may be working on some natural language processing projects now?
Kaylie: We can talk a little bit about the project we're working now.
Nidhi: Sure. So we're still in the brainstorming stages but probably going to be working on a project for class videos or lectures and basically analyzing students' responses or questions. And essentially pinpointing in the video exactly where that topic is discussed. That'll be beneficial for lectures, MOOCs, stuff like that, just online research.
Kaylie: I'm also in the same NLP class and we're contemplating a few different possibilities for our project but one of them is from x-rays that we see and we use to diagnose certain diseases. How would you, for example, produce a more contextual and informatie description of the diagnosis? And using that to better help the doctors in making their final diagnoses. So instead of just doing a binary label of "this disease is present, this disease is not" maybe the model would be able to generate a descriptional kind of outline of certain things to test for or reasons justifying its diagnosis. Also incorporating things like past patient records and medications they’re on, but all of this is also just in the early stages and we're still discussing with people who are collaborating with us and providing the data for us.
JM: Sounds interesting. How about you, Sarah any new things you're working on, bio related or bio-chemistry, deep learning, or computer science related?
Sarah: I'm in an interesting genomics class this quarter.
JM: Ah, wow. Okay. Great. I think that's about all I got. Thanks again for coming out and chatting with me, l’ll go ahead wrap it up here.

For this blog post, James Montantes from Exxact sat down with some Stanford CS students to discuss their project "Predicting Correctness of Protein Complex Binding Operations" that was presented at the CS229 poster session on December 2018. This particular poster session was of particular interest in that it shows an interesting application of deep learning for bio-chemistry, while comparing more traditional machine learning methods such as SVM to attack this particular problem. Students; Sarah Gurev, Nidhi Manoj, and Kaylie Zhu sat down with James for a quick 30-minute discussion at the bustling Coupa Cafe on the Stanford campus to discuss some of the technical challenges of the project.
James Montantes: Thanks for giving me the opportunity to talk about your project, it was very interesting. Typically, I go to these poster sessions whenever I get the chance, I think it's a great opportunity to see a lot of interesting applications for machine learning. Given its recent popularity in the past several years, there's some real industry use cases applying these algorithms to the GPU, solving real problems. At the university level, I find these poster sessions offer a lot of interesting use cases. Which brings me to your poster, I thought it was interesting because you're going after a unique application, which would be molecular dynamics. Given the field I’m working in, I felt it would be a great fit... Having said all that, tell me a little bit about yourselves and what you're working on, your background and interests etc.
Sarah: I'm a junior majoring in computer science, in the Biocomputation track. I've been interested in both biology and computer science for a while. I started off working in a pure bio lab doing cancer biology research, and then moved to a structural biology computational lab, the Levitt Lab, working on molecular dynamics which is how I was interested in our project. For lab, I'm modeling chemokine-chemokine receptor interaction using molecular dynamics and analyzing those to understand the basis of affinity of the interaction.
JM: Thank you. Kaylie?
Kaylie: Yeah, I’m a junior in the Symbolic systems major (AI concentration) and also co-term in CS with AI depth. I've been working previously with the Stanford AI group with deep learning applications regarding health care. Since then I've just been interested in deep learning and different kinds of applications in computer vision, for example, food recognition, satellite imagery, map technologies, et cetera. I really enjoy discovering new uses of deep learning and in this case, we've applied it to bio-molecular dynamics which is a new field for me and I thought that was very exciting.
JM: Ok, thank you, Nidhi?
Nidhi: I'm a senior studying computer science with a focus in AI. And, co-term student so it's the master's program here that’s also in CS with a focus in artificial intelligence, again. In terms of research interests, Kaylie and I were part of the same lab. That's how we actually met. It's the AI plus health care lab in the Stanford Machine Learning Group under Andrew Ng. And that is, as Kaylie articulated, deep learning, focused on health care problems, usually in radiology or pathology. Now I'm working with Fei-Fei Li in the AI plus health care sector of her lab that's also in the Stanford Machine Learning Group. During the summers I enjoy interning just to try to get a sense of both research and industry from both sides of it, to see the application side as well. And I guess, we were all kind of interested in the intersection of artificial intelligence and biology when we were taking a class together. All our backgrounds coming together was interesting to work on the bio-computation project.
JM: Great. Thank you, now this seems like a very specific problem, specific to finding protein docking orientations and what not… How did you come across this? How did you feel this problem could be solved with machine learning? And what was the inception of this idea?
Sarah: I talked to a postdoc in my lab, João Rodrigues, who’d made the HADDOCK models from Docking Benchmark data. So when I came to him looking for a dataset for CS229, he suggested this. It’s a good application for Machine Learning because there's so many protein complexes that we'll just never have their structures experimentally determined, but scientists would like to be able to use those protein complex structures to advance their understanding. So, it's useful to be able to use some techniques to determine what simulated models are the proper models.
JM: Excellent… So now going back to machine learning, what kind of machine learning, or deep learning tools or models did you use to solve your problems? How did that come about? I saw in your paper you used scikit-learn to use some of the regression testing for the SVM (Support Vector Machine) etc.
Nidhi: For the SVM implementation, which is our baseline for both regression and classification, we were using scikit-learn, which just really simplifies the implementation of the model. Then we transitioned into a deep learning, 3D CNN (Convolutional Neural Network) resident model that Kaylie can talk a little bit more about.
Kaylie: For the 3D CNN we used PyTorch, which is a super useful library for deep learning. All three of us prefer it over TensorFlow, so that's what we chose. Also, we built on top of a model that was built by other people for other uses called ResNeXt. It's a 3D convolutional neural network that's used for 3D data and commonly used for video recognition. In our case, it made more sense, because our cubes were three-dimensional and we needed to capture spatial information. It just works extremely well in capturing the spatial context of molecules and the atoms architecture. It was super convenient for us to build because this library has built-in ways to tune learning rates and batch sizes and we could experiment with different model depths; for example, we tried ResNet 50 & 101, and we found some interesting results.
JM: Ok, I know the ResNet is a very popular model, and it's widely used. So the ResNeXt is kind of a newer architecture?
Image source: Aggregated Residual Transformations for Deep Neural Networks, Saining Xie, et al. 2017
Nidhi: It's better for 3D data
JM: I see, it lends itself to 3D spatial data.
Nidhi: Yes, and just for context, basically what we're trying to say is the protein complex equals a 3D representation, that's why a 2D model probably wouldn't work as well. The main reason you use ResNet or ResNeXt is residual connections. It's just easier for training.
Kaylie: ResNeXt, it has the advantages carried over from ResNet of being highly modularized and skipping the layers in order to better perpetrate the network flow of information of the gradients. But it also has a new dimension called cardinality, basically it introduces group convolutions and controls the size of the set of more complex transformations, an essential factor in addition to (and in fact more effective than) the dimensions of depth and width (when it comes to gaining accuracy, as shown by our experiments).
JM: Do you know if there's a limit of how many dimensions it can spread out?
Kaylie: So ResNeXt is technically for 3D data, but people can enhance it in order to adapt for more numbers of dimensions as well. But for our case, it made sense to apply for 3D.
JM: When when you implement a large ResNet model or you're training on a huge data set, it can take a long time. How about your model and dataset using ResNeXt, does it take a long time to train? Was it very data intensive?
Sarah: The data input is relatively small compared to large images with many pixels. Our data was only 10X10X10. Each voxel of that is a 1X1X1 cubic angstrom box, about the size to indicate whether one atom was present. We split our protein into these 10X10X10 cubes along the interface between the two proteins and each voxel has a zero there no atom was present or else a specific number depending on the atom. This allowed us to have a 3D representation of the complex, but where inputs were all the same size.
Nidhi: Also, for context, yes, it does take a long time to train. We were using a GPU from the Stanford Levitt Lab, so that made it realistic to train under our time constraints.
Kaylie: And ResNeXt 101 was definitely slower to train than (ResNeXt) 50.
Sarah: We got our results within a day, so it wasn't too long, and was within the scope of our project.
JM: So you use this convolutional neural network, this interesting ResNeXt model, but you also tested it against the SVM regression and classification. Is that sort of a benchmark in that you wanted to see how well it performed against traditional machine learning algorithm? Was that idea behind it or you wanted to see "do we really need deep learning," or…?
Nidhi: So, usually, these projects, we want to start off with a baseline to build off of and build more complex models. For our baseline we decided to do SVM regression and classification as articulated in our paper. We chose those because we read in previous work that SVMs tend to have good results in this. And the features we used, also mentioned in our paper, were solvation energy, van der Waals, buried surface area, and electrostatics, that has been shown to have good results in the past. We started off with that, and then we made use of the computational power of deep learning networks and that's how we started working on 3D CNNs.
Kaylie: I think if you look at our paper, the accuracy of the training results are pretty high, but the tests were a lot lower. So that's basically saying that there is some significant amount of overfitting done by the SVMs. This was tackled a lot better in our 3D CNN model, which was able to learn the more complicated relationships within the dataset rather than just overfitting to the training data. We saw that after significant tuning, it was actually able to learn and generalize to the test sets. That was one of the significant improvements in using deep learning versus the traditional machine learning model. But sometimes, in other cases, traditional machine learning is enough to produce sufficient results. Just in this case, it wasn’t sufficient to capture all the information and data complexities as well.
Nidhi: Just for some context, usually in a machine learning project, you want to split up your data into training and test. So, you train essentially on the training set, and what Kaylie was saying is when you overfit, it's kind of understanding those examples ‘too well’, therefore when you give it new data in the test set, it's not able to generalize. Basically, the SVM model was a little too simple. It was just overfitting on the training set, so that's why we resorted to CNN, which had better results in the test set, which is what you want.
Sarah: It was important that we split by complexes when we were dividing our data into training, validation, and test sets. There's 10000 HADDOCK dockings for each of the 135 complexes. We split by complex, so that when we are testing our model, it hasn’t already been trained on simulations from the same complex, preventing bias.
Nidhi: You can think of it as our problem is essentially: if given a new protein complex, how would our model be able to perform on that? So that's why your performance in the test set, and making sure you have complexes that your attributes, after you've trained this, after you've built a great model, it can perform well.
JM: Okay. That's great. That's good information. You mentioned that you gave with your training data sets, kind of a more positive results? What was that?
Sarah: There are significantly more bad models than good models, so we needed to reduce the number of negative models. Because we do 10000 dockings for each complex and the orientation is random, only a few of them are going to be proper, and by proper, I mean the RMSD, the root-mean-square deviation from the true protein complex, is small. So, only a few of these random orientations will be close to true. We found it necessary to significantly undersample the bad examples so that we had a comparable number of good examples and bad examples to train with.
Nidhi: Think about how proteins could combine, there's like a million ways they could combine. The reason you want an equal number of positive and negative is just that will help a lot with training. When you have class imbalance between positive and negative samples, it's just a lot harder for the model to learn. Yeah.
JM: I see. Fair enough. I also noticed in your paper, you mentioned some existing tools that maybe don't specifically go after the same problem, like AtomNet, that's commercially available, is it not? I was just curious had you had any experience with any of these: There's ENZYNET, which is a two-layer 3D convolutional neural network for enzyme.
Sarah: We've never used any of these models, but we're encouraged to find background research so we can understand what makes our project different from what's been done in the past. We find the pros and cons of the methods that have been used before.
Nidhi: You look to see what's happened before and want to learn from that, and guide your future research, like what kind of models are you can use.
JM: I see some of the ones that you're using like SVM models, like ProQdoc, ZRANK.
Nidhi: Yeah.
Sarah: The past models helped us also choose our parameter values for the SVM, because we do a grid search over a specific range. So we also used that same range as the past models because they are likely to be appropriate.
JM: That makes sense. This ties into a question that maybe you could help with...let's say you’ve worked in Life Sciences for years and years and done a lot of molecular dynamics work, but maybe just hearing about some of this ‘deep learning stuff’, or don’t really know how to really approach or implement some of these experiments. What kind of advice would you give them? How would they basically want to get started in it? Please explain in your own words.
Sarah: If they have a specific project in mind it's definitely a good idea to see what's already been done. They can also repeat what's already been done just for themselves, so they can learn the steps of what to take, and use tools like scikit-learn to do a basic experiment and understand the process.
Kaylie: You can also look at what kind of evaluation metrics most people, or other published work in the field have been evaluating models on because that's important. What aspects of the model performance do people actually care about, or what does the industry actually care about. Also, I think, if they have less background, they could look into transfer learning, which is basically transferring learned knowledge from a network of a different but similar problem to a new problem, and that requires a lot less technical depth, but it sometimes actually does amazingly well with some type of parameter tuning. So, if they are interested in image recognition or image captioning. There's a lot of networks that have been built by other people and if they’re are similar enough and with pre-trained weights, then they could load pre-trained weights, then either modify the last few layers or just freeze or retrain layers in order to adapt it to their problem. At times, it works exceedingly well.
Nidhi: I would say, I think if you want to get more...If you're interested in ML and want to learn a little bit more, I would start off by taking Coursera classes or reading up on MOOCs
JM: (Somewhat jokingly): How about CS 229 at Stanford?
Nidhi: Exactly. But maybe something a little easier than 229.
Kaylie: (CS) 230
Nidhi: Yeah, 230, 229, they're all great. I think that provides some necessary foundation of underlying concepts that's necessary. Otherwise, I think, I guess jumping into research papers can sometimes be a little daunting and just a lot of technical terms and concepts that are a little hard to grasp without any previous understanding. Then I think once you read up on papers, just try working on some projects. Make use of libraries, like Sarah said. Machine learning is a community, so you want to build off of the amazing work that's been done before in the field for whatever you work on. Also, all the results you have, contribute it back so that other people can build off it and keep pushing the field forward.
JM: Yeah, I've noticed that myself with the machine learning community. A lot of the software, and a lot of the papers are all open for anyone to read and download and try the code.
Nidhi: Exactly. Open source code.
JM: It's not always like that with some other areas. I've seen that trend as well, it's really welcoming...Going back to the experiments, what was the most time-consuming process or what was the thing that really frustrated you the most in this?
Kaylie: Data Prep!
Sarah: There's a lot of data processing, to convert our starting data into a usable form that can go into a deep learning model. We started with a PDB file, which is basically just a list of positions for every atom.
Nidhi: Protein data bank.
Sarah: You have all the atoms and you have the position in 3D space of those atoms, and you want to somehow turn that giant list into a 3D representation of a small portion of that complex that represents the interface. So, the way I ended up doing this was looking for pairs of amino acids, where one was on one protein and one was on the other. I’d then find the center of mass of each of those pairs and clustering those center of masses. I’d take the center of each cluster to be the center of the cube entering our model and then building the cubes around that. It was just a lot of work that went into turning this list of all these atoms positions into a usable form.
JM: And then that would be interpreted by the neural network?
Sarah: Some tools that we used were PyMol for visualization and MDTraj. It was important that we randomly rotate each complex because we didn’t want a specific orientation to influence the model. Since we have multiple orientations that are put in the model for each complex, we don’t want all those orientations to be the same.
JM: When you're setting the parameters for the network, how did you go about that? Was it established on things that have been done before that you thought would work well? Was it a lot of trial and error? How did selecting those come about?
Kaylie: So, there's two parts to it. From related work you can kind of ball park where the hyper parameters would be and then narrow down by experience and empirically. For example, ADAM optimizer tends to do well with its default parameters, you don't usually need to tune it as it doesn't produce as much effect. I think the first thing to tune would be learning rate, we usually default with 1e−3. But for us, we were like, "Oh this is actually too big," it was very clear once you look at how the training error has been decreasing over the epochs, you're clearly seeing that it's actually missing the optimum point and it was jumping around a lot so we decided to decrease the initial learning rate.
JM: Ok, so don’t mess with ADAM optimizer, and tune the learning rate starting at 1e−3.
Kaylie: We also reduced learning rate on plateau, which is a very handy trick that we found to be useful in other projects as well. We just decided to use it basically once the validation loss plateaus, we reduce the learning rate in order to find more fine-grained way of finding the optimum point. And rather than risk either missing it or not reaching there in time.
Kaylie: Also, things that we try out are batch sizes, which is significant. There's a lot of empirical testing to it, for example, you usually try out 64, 32, 16, et cetera, depending on your memory constraints and what works best for the problem. I guess there's a lot of ‘rule of thumb’ you can follow and things you can learn from experience and also from other people’s work. But there's a lot of empirical testing, which I think is actually really inevitable in work like this.
JM: Excellent. Okay, so I think that pretty much goes over most of the technical questions. Was there anything else you want to add or points of interest? I know you may be working on some natural language processing projects now?
Kaylie: We can talk a little bit about the project we're working now.
Nidhi: Sure. So we're still in the brainstorming stages but probably going to be working on a project for class videos or lectures and basically analyzing students' responses or questions. And essentially pinpointing in the video exactly where that topic is discussed. That'll be beneficial for lectures, MOOCs, stuff like that, just online research.
Kaylie: I'm also in the same NLP class and we're contemplating a few different possibilities for our project but one of them is from x-rays that we see and we use to diagnose certain diseases. How would you, for example, produce a more contextual and informatie description of the diagnosis? And using that to better help the doctors in making their final diagnoses. So instead of just doing a binary label of "this disease is present, this disease is not" maybe the model would be able to generate a descriptional kind of outline of certain things to test for or reasons justifying its diagnosis. Also incorporating things like past patient records and medications they’re on, but all of this is also just in the early stages and we're still discussing with people who are collaborating with us and providing the data for us.
JM: Sounds interesting. How about you, Sarah any new things you're working on, bio related or bio-chemistry, deep learning, or computer science related?
Sarah: I'm in an interesting genomics class this quarter.
JM: Ah, wow. Okay. Great. I think that's about all I got. Thanks again for coming out and chatting with me, l’ll go ahead wrap it up here.
