Scientific Computing
Generative AI Protein Design After the Nobel Prize
April 10, 2026
9 min read


Generative AI has fundamentally shifted what's possible in protein engineering. Where structure prediction once defined the field's ambitions, the focus has moved decisively toward building functional proteins from scratch, conditioned on specific targets, with unprecedented precision and throughput.
Models like RFDiffusion and AlphaFold3 don't just predict how a protein folds. They generate novel sequences and structures optimized for binding, catalysis, or therapeutic effect. The design space is staggering, with 20 canonical amino acids and even a modest 100-residue sequence, there are more possible proteins than atoms in the observable universe. Generative AI is the only practical tool for navigating it.
This post covers where the field stands today, including the latest model capabilities, emerging applications, and what it takes computationally to run these workloads.
Proteins are essential biomolecules that perform many functions in living systems. What makes them compelling for AI-driven design is straightforward: how they fold determines their function. A 100-residue sequence has 20^100 possible variants. Evolution has sampled a tiny fraction of that space, while Generative AI can uncover the rest.
Generative models have fundamentally expanded what protein engineering can do. Rather than searching that space exhaustively or relying on evolutionary data alone, diffusion-based architectures like RFDiffusion can sample functional structures directly, conditioned on specific targets and constraints. The result is a design pipeline that proposes structures nature has never tried.
PDB Molecule of the Month (PDB ID:[ 7vcf, TOC-TIC supercomplex from Chlamydomonas algae.
What modern generative models can condition protein design on:
This shifts the bottleneck in protein engineering from algorithms to throughput. Iterating across large candidate libraries, running ensemble sampling, and validating designs in silico before wet lab synthesis all demand serious compute, the kind of workloads purpose-built HPC systems are designed for.
Diffusion models were proven in image generation. The same core mechanism, training a model to iteratively denoise from random noise toward a coherent output (images and now, 3D models), transfers directly to protein structure. Instead of pixels, the model operates on atomic coordinates.
RFDiffusion, released by the Baker Lab in 2023, was the first model to demonstrate this at scale for protein design. RFDiffusion3, published on BioRxiv on September 18, 2025, is a significant step forward.
The key advance is all-atom design. Earlier models worked at the residue level, building the protein backbone first and handling binding partners separately. That approach creates problems: misfit pockets, wrong orientations, unrealistic chemistry. RFDiffusion3 designs the protein and everything it interacts with, including ligands, nucleic acids, and small molecules, simultaneously at the atomic level. The result is more chemically realistic designs from the start.
What RFDiffusion3 adds over its predecessor:
For practitioners, that last point matters. More candidates, faster iteration, same hardware.
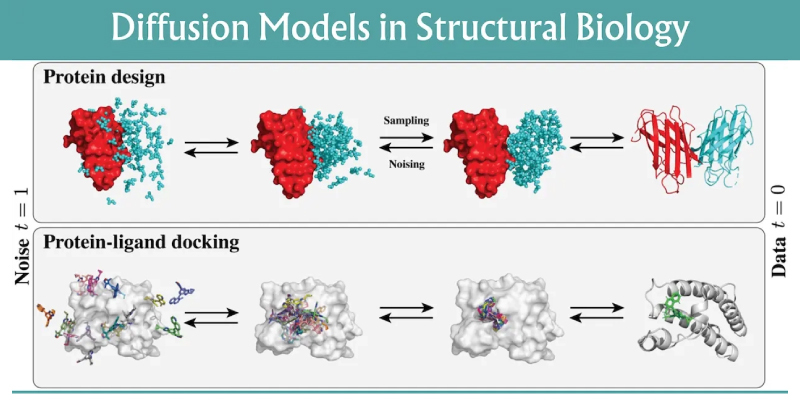
In a typical design workflow, each “run” is not a single inference. Teams often generate hundreds to thousands of candidates per target, filter them with fast scoring functions, then re-sample the most promising designs under tighter constraints, like a funnel.
Common stages in an AI protein design pipeline:
The compute challenge is that this pipeline is embarrassingly parallel, so teams naturally scale it out. That scaling quickly turns “a few GPU runs” into sustained multi-GPU throughput, plus the storage and orchestration to keep experiments reproducible.
Multiple GPUs running in parallel are the key to accelerating workflows. The more GPUs running, the more candidates a research organization can test and eliminate. High memory GPUs are also valuable to keep models on a single GPU. If models bleed into a secondary GPU’s memory, this can become a communication bottleneck.
The structure prediction problem is solved. The design problem is a different story. Generating a protein that folds correctly has largely been solved. Generating one that performs a specific function reliably is still hard. Very hard.
Current models have real limitations: accuracy drops for proteins over 400 residues, performance on flexible regions and multi-chain interfaces is inconsistent, and de novo function design outside of binding tasks remains an open research problem.
Active work is focused on:
For researchers working on these problems, the iteration cycle matters. Running experiments locally on purpose-built hardware removes the access and cost constraints of cloud infrastructure and keeps data in-house. The models are open, the tooling is mature, and the open problems are algorithmic. That means the researchers making progress are the ones who can run experiments fast.
The Nobel recognition accelerated attention to protein design, but the near-term impact is practical: more organizations are trying to operationalize design loops.
In all of these, the competitive edge comes from iteration speed: generating more candidates, learning faster from assays, and tightening the design loop.
Generative protein design is moving fast. The models are improving, the open problems are well-defined, and the gap between a computational design and a validated candidate is narrowing. Researchers and biotech teams looking to advance their work will need serious computing infrastructure.
At Exxact, we build and integrate configurable workstations, servers, and clusters for life science research. Whether you’re validating AI-folded proteins, exploring RFDiffusion locally, or running a full start-to-finish protein design workflow, Exxact works with you to scale computing at any stage.
We specialize in offering alternatives to cloud computing, which can snowball in cost as usage scales. An on-prem solution keeps sensitive research data local and lets teams iterate without incurring additional per-run costs. Reach out to discuss a GPU-accelerated system sized for your workloads.

With access to the highest-performing hardware and industry experts, Exxact offers platforms optimized for your Life Science reseach deployment, budget, and desired performance so you can continue your groundbreaking innovations.
Configure your Protein Folding System TodayGenerative AI has fundamentally shifted what's possible in protein engineering. Where structure prediction once defined the field's ambitions, the focus has moved decisively toward building functional proteins from scratch, conditioned on specific targets, with unprecedented precision and throughput.
Models like RFDiffusion and AlphaFold3 don't just predict how a protein folds. They generate novel sequences and structures optimized for binding, catalysis, or therapeutic effect. The design space is staggering, with 20 canonical amino acids and even a modest 100-residue sequence, there are more possible proteins than atoms in the observable universe. Generative AI is the only practical tool for navigating it.
This post covers where the field stands today, including the latest model capabilities, emerging applications, and what it takes computationally to run these workloads.
Proteins are essential biomolecules that perform many functions in living systems. What makes them compelling for AI-driven design is straightforward: how they fold determines their function. A 100-residue sequence has 20^100 possible variants. Evolution has sampled a tiny fraction of that space, while Generative AI can uncover the rest.
Generative models have fundamentally expanded what protein engineering can do. Rather than searching that space exhaustively or relying on evolutionary data alone, diffusion-based architectures like RFDiffusion can sample functional structures directly, conditioned on specific targets and constraints. The result is a design pipeline that proposes structures nature has never tried.
PDB Molecule of the Month (PDB ID:[ 7vcf, TOC-TIC supercomplex from Chlamydomonas algae.
What modern generative models can condition protein design on:
This shifts the bottleneck in protein engineering from algorithms to throughput. Iterating across large candidate libraries, running ensemble sampling, and validating designs in silico before wet lab synthesis all demand serious compute, the kind of workloads purpose-built HPC systems are designed for.
Diffusion models were proven in image generation. The same core mechanism, training a model to iteratively denoise from random noise toward a coherent output (images and now, 3D models), transfers directly to protein structure. Instead of pixels, the model operates on atomic coordinates.
RFDiffusion, released by the Baker Lab in 2023, was the first model to demonstrate this at scale for protein design. RFDiffusion3, published on BioRxiv on September 18, 2025, is a significant step forward.
The key advance is all-atom design. Earlier models worked at the residue level, building the protein backbone first and handling binding partners separately. That approach creates problems: misfit pockets, wrong orientations, unrealistic chemistry. RFDiffusion3 designs the protein and everything it interacts with, including ligands, nucleic acids, and small molecules, simultaneously at the atomic level. The result is more chemically realistic designs from the start.
What RFDiffusion3 adds over its predecessor:
For practitioners, that last point matters. More candidates, faster iteration, same hardware.
In a typical design workflow, each “run” is not a single inference. Teams often generate hundreds to thousands of candidates per target, filter them with fast scoring functions, then re-sample the most promising designs under tighter constraints, like a funnel.
Common stages in an AI protein design pipeline:
The compute challenge is that this pipeline is embarrassingly parallel, so teams naturally scale it out. That scaling quickly turns “a few GPU runs” into sustained multi-GPU throughput, plus the storage and orchestration to keep experiments reproducible.
Multiple GPUs running in parallel are the key to accelerating workflows. The more GPUs running, the more candidates a research organization can test and eliminate. High memory GPUs are also valuable to keep models on a single GPU. If models bleed into a secondary GPU’s memory, this can become a communication bottleneck.
The structure prediction problem is solved. The design problem is a different story. Generating a protein that folds correctly has largely been solved. Generating one that performs a specific function reliably is still hard. Very hard.
Current models have real limitations: accuracy drops for proteins over 400 residues, performance on flexible regions and multi-chain interfaces is inconsistent, and de novo function design outside of binding tasks remains an open research problem.
Active work is focused on:
For researchers working on these problems, the iteration cycle matters. Running experiments locally on purpose-built hardware removes the access and cost constraints of cloud infrastructure and keeps data in-house. The models are open, the tooling is mature, and the open problems are algorithmic. That means the researchers making progress are the ones who can run experiments fast.
The Nobel recognition accelerated attention to protein design, but the near-term impact is practical: more organizations are trying to operationalize design loops.
In all of these, the competitive edge comes from iteration speed: generating more candidates, learning faster from assays, and tightening the design loop.
Generative protein design is moving fast. The models are improving, the open problems are well-defined, and the gap between a computational design and a validated candidate is narrowing. Researchers and biotech teams looking to advance their work will need serious computing infrastructure.
At Exxact, we build and integrate configurable workstations, servers, and clusters for life science research. Whether you’re validating AI-folded proteins, exploring RFDiffusion locally, or running a full start-to-finish protein design workflow, Exxact works with you to scale computing at any stage.
We specialize in offering alternatives to cloud computing, which can snowball in cost as usage scales. An on-prem solution keeps sensitive research data local and lets teams iterate without incurring additional per-run costs. Reach out to discuss a GPU-accelerated system sized for your workloads.
With access to the highest-performing hardware and industry experts, Exxact offers platforms optimized for your Life Science reseach deployment, budget, and desired performance so you can continue your groundbreaking innovations.
Configure your Protein Folding System Today